在第一章中,我认为相似结构和DNA序列并不足以证明我们与黑猩猩有着共同的祖先。我检视经过同行审核的有关过渡型化石的科学文献,以及关于当前黑猩猩和人体解剖学的最新数据,我的结论是, 新达尔文主义的进化过程不可能完成我们与黑猩猩最后共同祖先之间应有的转变, 因为有太多结构上的变化, 并且时间太短了。
但是,关于人类起源目前的挑战不仅仅涉及化石,解剖学,还涉及达尔文主义的进化的不可能性。现在DNA测序已经变得相对简单和便宜,研究人员正在收集大量的人类基因组序列数据。他们使用遗传变异的发现重建在我们过去的基因历史事件,绘制进化树,估计祖先的人口数量,甚至计算出何时何地我们的祖先走出非洲。基于这方面的数据,有些人认为,人类不可能来自只有一对祖先夫妇。
这种说法直接与许多基督徒的传统信仰发生冲突,他们相信人类始自原始的一对夫妇--亚当和夏娃。那些隶属于像BioLogos基金团体的人更说,基督徒必须放弃以亚当和夏娃是人类唯一的祖先父母的信仰,因为据说科学论据已经证明他们是不可能存在的。
我是一个科学家,不是神学家,但我觉得有义务说话。造成对两个始祖的挑战是科学的数据,所以它值得科学的回应。我本章的目的不是为了解释圣经, 或要判断各种对亚当和夏娃持不同意见的基督徒的正确性。相反地,我将把重点放在科学的论证和它的有效性方面。
挑战亚当和夏娃群体遗传学的论点有多种形式。在这里我将研究对第一对夫妇挑战最强的案例之一---基于人类白血球细胞抗原(HLA)的基因论证,这个基因是一些在人类基因组中最可变的基因遗传变异。当我开始这项研究时愿意接受人类祖先可能多过两个人,因为人类有太多的遗传基因多样性,不可能只通过两名家长遗传至今。但我的发现令我惊讶,即使这个我们的最具多态性(最多样的)基因组也并不排除有第一对夫妇的可能性。而且我发现甚至埋在这个基因组中的证据有更多的东西, 表明我们的基因组成并非简单地从与其他物种的共同祖先遗传而来。
这里的科学数据是复杂的。为了认真评估所提出的论点,我不得不包括了相当数量的技术讨论。我意识到这章的某些部分可能对一些读者有挑战性,但我尽量在整章中以非技术的语言说清楚我的主要观点。
HLA的基因论证
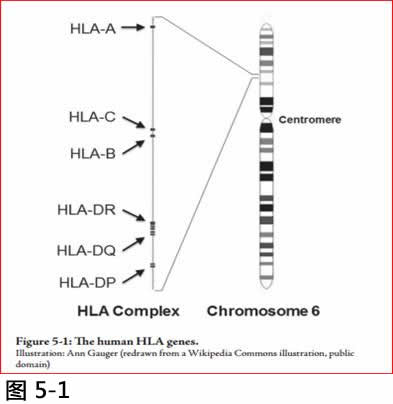
HLA基因(按:HUMAN LEUKOCYTE ANTIGEN:人类白血球抗原)参与免疫防御---它们与外源肽(foreign peptide)结合, 显示在免疫细胞(白血球)表面上, 以触发其他免疫细胞的响应。在哺乳动物中有很多这些HLA基因,大概是为了提供对抗多种疾病和寄生虫的免疫作用。图5-1显示在人体中的主要HLA基因的位置。
图5.1 HLA基因
Illustration: Ann Gauger (redrawn from a Wikipedia Commons illustration, public domain).
目前已知的每个HLA基因有很多版本(等位基因)。正因为如此, HLA的结构, 是对我们来自只是两个第一父母的想法的极端恶劣的挑战之一,如果有数以百计的HLA等位基因存在于现在人口中,它们是从哪里来的呢?两个人可以最多有四个版本的传递。难道所有这些等位基因仅来自两个人所有的四个或更少的祖先版本吗?
要回答这个问题,我需要解释一些有关在这些研究中所使用的方法,以及他们潜在的假设。
什么是群体遗传学
20世纪30年代和40年代,达尔文的进化论和遗传学孟德尔理论相互结合,创造了现在所谓的现代综合论,我称它为新达尔文主义。新达尔文主义开始注重变异基因如何通过人群来传播, 而不是集中于动物如何随着时间可能有不同形式的演变, 这些被称为"群体遗传学家"的人开发数学模型,要从现有群体的遗传变异,来推断在过去这些人口可能发生了什么事。
因为所有这些模型都来自他们的达尔文主义的根源,所以他们认为自然选择作用在随机过程(stochastic process即随机发生的过程,并没有考虑生物本身的的需要), 就足以说明一切进化改变。产生遗传变异的随机过程包括突变(改变DNA序列)和基因重组(重排染色体之间遗传信息的交换), 基因漂流(由于生育故障遗传信息的随机丢失)有降低自然选择驱动遗传变化力量的趋势,特别是在一百万以下的人口。
请注意, 在新达尔文主义中不可能容许有方向或指导的进化。随机的基因变异在一次偶然的机会中发生, 没有顾念到群体的任何需要。自然选择会加以筛选,基因漂流抛出更多的随机性, 选择哪个变种基因可以实际生存和在人群中传播。
群体遗传学需要简化某些数学方程式才能运作。大多数模型使用目前的遗传多样性,以模拟追溯过去的事件, 它们假定不变的背景突变率,没有强大的被选择偏向的遗传变化。它们假定一个恒定的群众数目, 无群体的迁移,和它们假定共同祖先是DNA序列相似性的根本原因。所有这些假设都受到质疑,且看下文的分析。
群体遗传学挑战第一对人类的祖先
在20世纪90年代的名为弗朗西斯科·阿亚拉 (Francisco Ayala)的群体生物学家采用从HLA序列得来的遗传信息, 开始着手挑战两个人的第一对人类祖先的思想。(注1) 阿亚拉选择了HLA-DRB1的一个原因,是那个时候有已经有数百已知的HLA-DRB1不同的版本。因此他有理由怀疑在假想当黑猩猩和人类的谱系分歧进化时,在HLA-DRB1序列可能有已经相当大的差异。
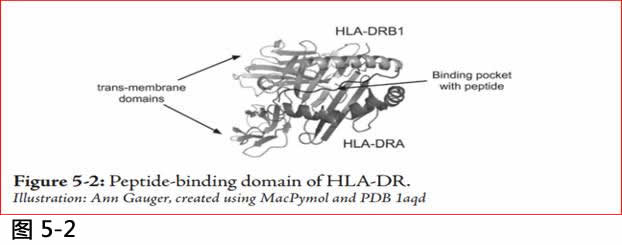
图5.2 : HLA-DR的肽连接结构域 (peptide binding domain)
Illustration: Ann Gauger, created using MacPymol and PDB 1aqd.
HLA-DRB1有什么功能呢? 为什么会有这样多的变异?HLA-DRB1蛋白与另一蛋白HLA-DRA结合,形成称为二聚体蛋白的HLA-DR(参考5.2)。(二聚体dimer是一个由两亚基蛋白质subunit组成的蛋白质) 。这个蛋白二聚体嵌入在抗原呈递细胞 (在免疫系统中特定类型的细胞) 的细胞膜上。该二聚体形成结合外源肽的肽结合袋,并将它们呈现给其他的免疫细胞,以触发它们生产合适的抗体。
为什么有HLA-DRB1的那么多的变异呢? 原因是,在肽结合袋的许多变化,可确保许多不同的外源肽被识别和结合。这是一件好事,因为它能够增强免疫力。如果一个新的寄生虫或致病微生物出现时, 群体中某些个人将有HLA-DRB1的基因, 能够结合入侵者"被提炼过而成的"外源肽蛋白质,并引发免疫系统启动一个防御它们的部位。
这里可见到有趣的事情: 几乎所有的遗传变异, 都是位于被用来绑定多变化外源肽的HLA-DR二聚体, 它们只是来自一个HLA-DRB1中的部分专门基因,即外显子(exon)2。(注2) HLA-DRB1或HLA-DRA的其余基因部分变化不大(按: 外显子乃是被转译为蛋白质的基因序列)。
阿亚拉获得黑猩猩、人和猕猴只从HLA-DRB1的外显子2 取来的DNA序列,使用群体遗传学算法重建那些序列的系统发展历史。 (注3) 他用最紧密贴合外显子2遗传变异模式的方法, 画了一个进化树, 又使用其他来源估计的平均突变率, 和黑猩猩和人类最后共同祖先的估计时候,他计算出共同的祖先在他树上的分歧点有多远。在此一分歧点绘制一个分界线,他数算人类有多少祖先的分支交叉。这样他给黑猩猩/人类最后的共同祖先, 在人群中应有多少HLA- DRB1基因, 作了回顾性的评估。 (注4)
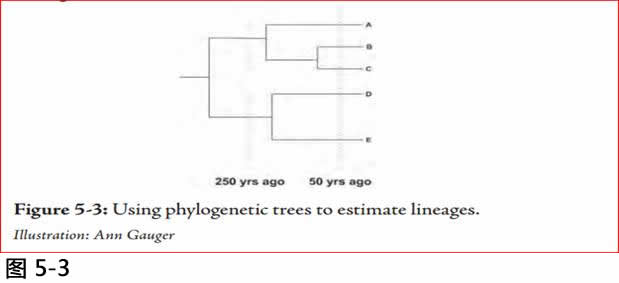
为了说明他遵循的基本过程,我画了一个进化基因树(图5-3)一个简单的例子。左边是树的最古老的部分。随着时间的迁移,单基因的重复和发散,然后再分枝数次。右侧最终分枝的数目为五。(A-E)
图5.3: 使用种系树来估计谱系。
Illustration: Ann Gauger.
通常群体遗传学家使每条种系树水平线的长度, 与遗传改变的量成正比例。长度越长,核苷酸差异越多。假设核苷酸差异是由于突变随着时间发生, 并假设突变发生的速率是恒定的(这并不是肯定的事), 人们可以向后倒数到在进化史的估计时间(在这种情况下,50和250年前), 并通过画一条与树垂直的线, 线穿过种系树的交叉, 确定在每一个特定的时间中出现多少独立的谱系。如果所有的估计是正确的,这棵树50年前有五个谱系, 250年前有两个谱系。
根据这一程序, 阿亚拉计算, 在我们最后与黑猩猩共同祖先的估计时间, 是四百万至六百万年前(并非肯定的事, 这些估计数字不断变化), 群体中HLA-DRB1基因共有32个分别的版本。为了让所有这些基因变异可以遗传到现代,他还估计祖先人数的最低不少于4,000,长期平均有效群体的大小为100,000。 (注5) 像阿亚拉的稳定状态的人口模型, 这个大数位是必要的。在这种条件下,假设随机交配和基因漂流,随着时间的推移, 基因是容易丢失的,这样一个大的始创族群是必要的,可以保证所有的基因继续传播。因为这个最小估计是4,000,阿亚拉声称,人类群体在任何时间内, 都不可能只通过两个人的基因瓶颈遗传而来。在他看来,在HLA-DRB1的变异实在有太多祖先的多样性。
对挑战的挑战
让我们回来检查阿亚拉的分析是怎么做的。他反对亚当和夏娃的两人袓先谱系, 是基于群体遗传学模拟随着时间而来的人群基因变化频率,以及祖先基因谱系如何易于凝聚在一起。他用来重建这些谱系树并计算祖先族群大小的方程式有赖于简化和假设,使数学可以运作,正如我前面所言。这些明确的假设包括在时间过程中有一个恒定背景突变率, 正在研究的DNA序列中缺乏遗传基因的拣选,群体的成员随意交配, 繁殖的群体大小没有因移民增加或缩小,并有恒定的群体人口。如果这些假设变得不切实际的话,这模拟的结果可能存有严重的缺陷。
群体遗传学模型中也有埋着隐藏的假设, 是被依赖来证明它们的结论,例如,谱系树的绘制法假定共同祖先树的存在。群体遗传学公式还假设随机过程是遗传变化在历史中发生的唯一原因,这是从自然主义得来的一个假设。如果有非自然的因素,甚至是未知的自然原因, 不按着随机过程进行干预而产生的遗传变化, 就不是在它的解释范围之内。
事实证明,从阿亚拉用于他的分析的特定DNA序列HLA-DRB1,是保证会获得高估的数据,因为他没有充分安排两个上述的假设 --- 假设正在研究的DNA序列中缺乏遗传基因的拣选, 和在时间过程中有一个恒定背景突变率。 HLA-DRB1被称为是有被强烈选择的异合子性能,这意味着有两个不同版本的基因给你一个更好的机会对付寄生虫和疾病。不仅如此,阿亚拉研究的特定区域(外显子2)似乎有一个比背景突变率要高得多的突变率。事实上,它是在我们的基因组的最可变基因之中的最可变区, 并且它可能是一个基因转变的热点(hotspot for gene conversion)这种突变特别可能混淆以共同祖先和简约系列为假设的谱系树绘制法), 我们在下文会讨论此事。阿亚拉确实为第一个问题使用了一个数学修正子数,但是他并没有修正第二个问题。
通过贝里斯特伦等(Bergstro?m et al.)(注6)检测同一HLA-DRB1基因后来的研究, 获得很不同的结果, 所用的基因不能够转译成蛋白质的内含子2(intron 2)。他们选择这个与外显子2毗邻的一个内含子序列, 明显地是要避免强的选择和一个高突变率,和/或基因转换的混淆影响。他们验证了这一内含子的突变率, 与基因组的背景突变率接近。这项研究得出的结论与阿亚拉的研究相异, 是在四百万至六百万年前我们最后与黑猩的共同祖先世代,只有七个版本的HLA-DRB1基因, 而那时长期平均有效群体的大小是10,000,而不是阿亚拉估计的100,000。
换句话说,通过对只是上面假设中的两个结果密切注意,这些研究人员在祖先人口中获到的估计基因数量, 比阿亚拉在他的研究中发现的数量显著地降低(即从32个HLA- DRB1基因数目降低为7个)。但是阿亚拉的模式有更深的问题,我们将在下一节中看到。
种系混淆(Phylogenetic Confusion)
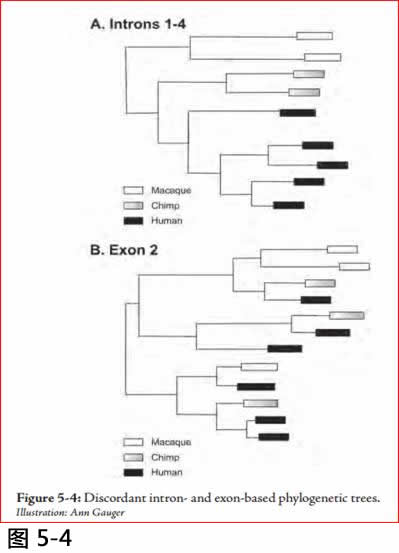
阿亚拉以外显子的HLA-DRB1基因2序列创造了他的进化树, 而贝里斯特伦等人却使用内含子2序列。第三个,多斯亚狄斯Doxiadis 等的研究再次检查黑猩猩、猕猴和人HLA -DRB1基因的种系历史,但这次他们采用的数据, 是从外显子2或内含子1-4取来的序列。出人意料的是,采用外显子2与使用内含子1-4绘成的进化树互相比对之下, 给基因的进化历史带来显著不同的系列,尽管这两组序列都来自于同样的基因。其中有一个在亲缘关系上存在实质的差异: 外显子2比较典型地表现出跨种间的关联,而内含子比较发现种与种之间的种内关联 (注7)。
不和谐种系树的简化示意图示于下列图5.4。 (有关原来的种系树,请参考多斯亚狄斯等,注8)。内含子序列组很明显地是根据品种分布的,而外显子2序列显示没有遵循物种而来的关系。
图5.4 :以内含子和外显子为基础的不协调的进化树。
Illustration: Ann Gauger.
虽然基于基因的进化树有时比较不显示物种之间的亲缘关系, 正如外显子2序列的情况。但是这种情况应该是令人惊讶的, 它表示发生了不同寻常的事情。
更不寻常的是, 用来自同一个基因的相邻节段所绘制的种系树彼此矛盾。它不是因为外显子2是高度可变的, 以及内含子是更保守的。内含子谱系相互之间可以有很多不同的地方。不寻常的是,内含子谱系组都是根据品种排列,而外显子2系别却不是。
一些进化生物学家尝试解释HLA-DRB1种系树的不协调。他们争辩说, 这证明这些基因始源在黑猩猩、人类和猕猴的谱系分离以前的深远进化历史,它是外显子2的数据所定义的基因种系历史。(注9) 还有人认为历史中出现了在跨物种不同的外显子2序列之间, 古迹肽结合基因序列发生改组,但在内含子谱系却不变。 (注10),但是目前尚不清楚,外显子2的这种跨物种分类的序列拼凑, 如何可能不破坏旁边在物种持续的内含子。此外,这将要求初始物种种群, 在漫长的时间周期中混杂在一起。混杂时间周期不太可能持续三千万年之久,因为这是最后一次猕猴、黑猩猩和人类理应拥有共同的祖先的时期。而且外显子分支长度可以媲美有种内关联内含子序列种系树的分支长度, 或是比它更长的事实,表示其中许多内含子谱系已经独立发展了相当长的一段时间,有些很可能已超过三四千万年。因此这个HLA-DRB1种系树的不协调, 不能由共同的祖先来解释,特别是当我们考虑额外的一项数据: 第6染色体的HLA-DRB1区域显示很少重组的迹象。
特殊行为,特殊的设计?
HLA-DRB1最亲密的邻居是 HLA-DQB和HLA-DQA ,也是与外源肽结合并展现给其他的免疫细胞,正如HLA-DRB1一样。根据雷蒙德等人的研究,这个区域显示了极端的连环不平衡(linkage disequilibrium,即在不同位置的基因一同遗传),这意味着这些基因之间几乎没有相互重组过。 (注11)
缺乏重组是非常不寻常的,因为它扩展了超过80,000个碱基的DNA。(注12) 绵延不发生基因重组的基因序列被称为单倍型(haplotype)。在通常情况下若考虑到这些单倍型的假想年龄,重组应该大约在每150个核苷酸之间发生。重组确实在该地区的其他地方发生,只是没有发生在HLA-DRB1附近。
尽管每个HLA基因有数百个等位基因,仅是某些HLA-DQ和HLA-DR的等位基因组合有一起遗传的趋势--它们是一块儿被遗传。这可能是因为这些特定等位基因的组合具有尤其好的合作效率,而其他不太有利的组合, 通过自然选择在人口中被删除。另一解释是重组可被一些其他机制来抑制。
共同遗传的等位基因组合构成HLA-DRB1的基本单倍型。大多数研究人员都同意,在现代人口中只有五个这些基本的单倍型。其中HLA-DRB1基因的某一特殊单倍型, 会具有与其他特定等位基因的组合。根据在内含子遗传背景的变化量,三个单倍型似乎是古老的,有三千万年以上的年纪。这是我们与黑猩猩和猕猴共有的单倍型。基于其积累背景突变,其余两个单倍型更年轻,可追溯到约四百万至六百万年以前。(注13)我们若在不同的历史时间中假设人与黑猩猩和猕猴分歧进化, 古人类分化时有可能最少至三个祖先单倍型, 或最多达到五个。
结论
下面是用简单的语言整理的结论。群体遗传学的论点是有太多的遗传多样性,不可能只借两个人一个瓶颈,即是亚当和夏娃一对夫妇的后代繁殖而来。但是事实证明是这理论是不正确的。
事实上,若引用所有的数据,目前只有五个基本的HLA单倍型的版本。三个似乎是古老的,要早于黑猩猩和人类之间的假想进化分裂时期,两个是比较近期的(若在灵长类动物和人类的假定最近共有始祖进化分裂时期的前后,这在于你在谱系树上哪里画线)。其中至少有这一个单倍型在黑猩猩中丢失。由于HLA区域的不寻常的遗传特性, 与估计谱系树上种系分歧时间的困难, 其中四个或更少的HLA单倍型可能是早于黑猩猩和人类之间的假想进化分裂时期。
每个人携带HLA单倍型的两个副本,所以每个人可以携带HLA-DRB1两个不同的等位基因。所以这四个单倍型可能是由只有两个人繁殖遗传而来。这是指出第一对夫妇可能有充分的遗传多样性来解释四个古基本单倍型,特别是考虑到人口迅速膨胀的可能性之后。
我们从基于DRB1外显子2进行审核,估计到有32个系列版本, 若使用DRB1内含子2数目下降成为7系,然后当考究整个HLA区域的序列时, 更减致3至5个祖先单倍型。这是一个了不起的逆转。曾经似乎是对的第一对夫妇的存在坚如磐石的论据, 现在已经大大缩小。这些遗传分析表明,第一对夫妇的存在是可能的。至少人们可公平地说,HLA单倍型多样性不能排除两个第一代父母的可能性。
那么究竟我们应该如何处理基因漂流的问题,以及随之而来所需要大量的人口(按:长期平均有效群体的大小),以防止变种单倍型的流失?这个问题只适用于一个固定的人口数量的稳定状态模型,而不是应用在人口迅速增长的情况。在一个新兴(创建)物种的情况下,迅速膨胀会使它保留所有单倍型。事实上有证据表明,一个新的种群成立之后HLA的多样性迅速增加,虽然通常没有快到这种程度。(注14)
现在我想向一个更具挑战性的方向发展。如果我们的DNA序列相似性不是源于共同祖先,其结果又怎么样呢?我们可能有两个源于智慧设计的始祖吗?在我已经提出的数据中有没有任何证据表明这个可能性?如果是这样,所有这些分析,我们与黑猩猩有多少古老共享单倍型其实就都不重要了。
HLA-DRB1基因中的当然有奇怪的变异图案, 表明可能有未知的进程在运行。我认为这个进程产生外显子2特异性高的可变性, 和禁止其他地方的重组。这个过程是针对在肽结合结构域要产生多样性。我认为为了要迅速产生新物种基础后的HLA多样性(假设我们来自两个第一的父母), 聪明的设计必须始自参与之初。支持这个想法的证据来自HLA-DRB1的多元性, 没有人会否认这个基因从少数的变异在六百万年以内演变成为超过六百个等位基因的事实。此外若比较其周围的DNA序列,在HLA-DRB1 2号外显子的可变区表现出错落杂拼和跨物种的关系, 很难被共同祖先的遗传来解释。来自不同物种相似图案的反复使用可能表明智能的设计。我还认为该过程是人类特有的,因为其他灵长类动物谱系内显示的等位基因多样性程度, 不能与人类系列相比。(注15)
这个提案至少部分地被已公布的数据所支持。基因转换(gene conversion)和超常突变(hypermutation)这两个已知的遗传功能会在其他免疫细胞基因系列中产生多样性的变异。(注16) HLA-DRB1基因序列的分析表明,"无论是在只有2号外显子的序列中或涉及相邻的内含子基因都有重组事件发生",和"指示在谱系之间的重组事件可能被隐藏,并且可能比目前预期的更频繁"。(注17) 另一些研究确定被认为参与在重组过程的序列特征,其中一些跨种族却高度相似的HLA-DRB1等位基因(注18)。
此外,几个人口研究的报告表明,许多的HLA-I类和II类基因经历快速等位基因之间的重组(interallelic recombination)。例如赫德里克和金报告:
在南美印第安人群中已发现新的等位基因, 这基因似乎是与其他等位基因之间微重组(interallelic microrecombination)的结果。由于美洲的移民可能只是来自一万至二万年前(约一千个世代以前), 这个不出现于亚洲人群中的新变种,必须在这一时期中出现。 (注19)
这些基因包括HLA-DRB1, HLA-DPB1,和HLA-B一些新的变种。(注20) 赫德里克和金继续说:
有直接的证据表明在某些MHC基因序列(按:即HLA基因所属的系列)中微重组频率高……根据赞根伯格等人(1995)的研究。在异合子基因型男性精子在HLA-DPB1基因高度可变外显子2的六个序列, 在111675个精子中, 有9个与其他等位基因之间基因转化,即是他们观察到近万分之一的配子有这个改变。(注21)
有鉴于这种数据,下列的提议似乎并非不合理: HLA-DRB1的多元性, 是一个过程的产品。这个过程在外显子2中引起特定的超变异和/或基因转换,以便迅速地产生HLA的分歧。这一个过程的存在, 基本上拆除了任何群体遗传学用以左右古代人口数目的数据。
这个HLA故事很好地说明了科学的优点和局限性。科学的声称是临时的,总是在被修订的过程中。特别是在追溯以往事件的计算时应谨慎,因为涉及未知的变数和隐藏的假设。其中涉及古代遗传史时,教条式的声明是不恰当的。我们对我们自己的基因结构知道得太少,使我们不能在过去遥远的遗传史上作精确的计算。但是,我们仍然发现很多有趣的事情,要考虑新的提案。
重新考虑进化故事
我选择研究HLA-DRB1的故事,因为它似乎是群体遗传学对两个第一父母的理论提出最强大的挑战,如果真的是我们与黑猩猩分享32个分歧的HLA-DRB1谱系,这个数据的确会造成一个原始的夫妇祖宗的困难。但是,正如我们上文所看到的数据表明,我们能够来自只有两个人的第一个祖先。
此外,数据表明DNA相似度不会是一个简单的故事。我们发现人类DNA有比黑猩猩更接近于大猩猩的序列。(注22),现在我们已经有更似猕猴的DNA序列(猕猴是灵长类却是不属于原始人组)。此外,当相邻的DNA区域产生不同的进化树时, 早早在黑猩猩和人类的假定最近的共同祖先时期之前, 物种已经分歧进化, 一些不寻常的事情正在进行。
这结果带给我一个惊喜,使我重新考虑我们从类猿祖先共同的血统进化而来的整个故事。从我自己的研究, 我已经知道形式或结构的相似性, 不足以证明新达尔文主义共同祖先理论是可能的。我知道真正的蛋白质的功能革新,是超越自然的过程所能达到的。因此,我开始重新审视一切我所知有关人类起源的数据。我考查研究古人类学、进化心理学和群体遗传学的研究论文,我考查研究畅销书和教科书。我应用严密的逻辑来分析我们从类人猿进化而来的故事。
虽然我一直怀疑人类由新达尔文主义的机制进化的合理性, 由于这一切的阅读和思考的结果,我现在也怀疑共同祖先的理论是否恰当。
目前,新达尔文主义对人类来源的解释被普遍接受。但可能的是,当我们继续研究我们自己的基因组时,达尔文解释我们与黑猩猩的相似性--即共同祖先--会被推翻。我们可以发现在我们的基因组的附加功能,轻看基于共同祖先的解释。当共同祖先的证据不足时, 我们将需要发展和进行测试可以替代的方案。
但是现在有一论点是清楚的:科学证据都没有显示亚当和夏娃的不存在,如果谁作如此的声称,他们就是歪曲了科学证据。
注释:
1. Ayala was not the only one to do this. See N. Takahata , "Allelic Genealogy and human evolution," Mol Biol Evol 10 (1993): 2-22.
2. Briefly, HLA-DRB1 has six exons (the coding regions) interspersed by noncoding DNA, called introns.
3. Phylogenetics is the study of evolutionary relationships among organisms.These relationships are often represented as branching trees. Starting with the assumption that common descent is true, scientists compare the distribution of varying anatomical traits or DNA sequences that they are studying. Usingmathematical algorithms, they look for tree-branching patterns that minimize conflict, or represent the fewest changes over time, but that can explain the observed distribution of traits or DNA variation.
4. Francisco Ayala, "The myth of Eve: Molecular biology and human origins," Science 270 (1995): 1930-1936.
5. H. A. Erlich et al., "HLA sequence polymorphism and the origin of humans," Science 274 (1996): 1552-1554.
6. T. F. Bergstr?m et al., "Recent origin of HLA-DRB1 alleles and implications for human evolution," Nature Genetics 18 (1998): 237-242.
7. G. Doxiadis et al., "Reshuffling of ancient peptide binding motifs between HLA-DRB multigene family members: Old wine served in new skins," Molecular Immunology 45 (2008): 2743-2751.
8. Ibid.
9. J. Klein, A. Sato, and N. Nikolaidis, "MHC, TSP, and the Origin of Species: From Immunogenetics to Evolutionary Genetics," Annu. Rev. Genet. 41 (2007): 281-304.
10. Doxiadis, "Reshuffling of ancient peptide binding motifs."
11. C.K. Raymond et al., "Ancient haplotypes of the HLA Class II region," Genome Research 15 (2005): 1250-1257.
12. There is an illustration of HLA-DRB1 and its neighboring genes in C. K. Raymond et al., "Ancient haplotypes," 1251.
13. G. Andersson, "Evolution of the human HLA-DR region," Frontiers in Bioscience 3 (1998): d739-745.
14. V. Vincek, et al., "How Large Was the Founding Population of Darwin's Finches?" Proc. R. Soc. London Ser. B 264 (1997): 111-118.
15. G. Doxiadis et al., "Extensive DRB region diversity in cynomolgus macaques: recombination as a driving force," Immunogenetics 62 (2010): 137-147.
16. Ziqiang Li, Caroline J. Woo, Maria D. Iglesias-Ussel, et al.,"The generation of antibody diversity through hypermutation and class switch recombination," Genes Dev. 18 (2004): 1-11.
17. Katja Kotsch and Rainer Blasczy, "Interlineage Recombinations as a Mechanism of The Noncoding Regions of HLA-DRB Uncover HLA Diversification," J Immunol 165 (2000): 5664-5670.
18. Jenny von Salomé and Jyrki P Kukkonen, "Sequence features of HLA-DRB1 locus define putative basis for gene conversion and point mutations," BMC Genomics 9 (2008): 228, accessed March 6, 2012, doi:10.1186/1471-2164-9-228.
19. P. W. Hedrick and T. Kim, "Genetics of Complex Polymorphisms: Parasites and Maintenance of the Major Histocompability Complex Variation," in R. S. Singh and C. B. Crimbas, editors, Evolutionary Genetics: from Molecules to Morphology (New York: Cambridge University Press, 2000), 211-212.
20. E. A. Titus-Trachtenberg, et al., "Analysis of HLA Class 11 Haplotypes in the Cayapa Indians of Ecuador: A Novel DRB1 Allele Reveals Evidence for Convergent Evolution and Balancing Selection at Position 86," Am. J. Hum. Genet. 55 (1994):160-167.
21. Hedrick and Kim, "Genetics of Complex Polymorphisms"; Gabriele Zangenberg, et al., "New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm," Nature Genetics 10 (1995): 407-414, accessed March 6, 2012, doi:10.1038/ng0895-407.
22. A. Hobolth, O. F. Christensen, T. Mailund, M. H. Schierup, "Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model," PLoS Genet 3 (2007): e7, accessed March 6, 2012,doi:10.1371/journal.pgen.0030007.